
{kind=link}

[ad_1]
Whereas vital strides have been made in predicting static protein constructions, understanding protein dynamics, influenced by ligands, is crucial for greedy protein perform and advancing drug discovery. Conventional docking strategies usually deal with proteins as inflexible, limiting their accuracy. Though molecular dynamics simulations can recommend related protein conformations, they’re computationally intensive. Latest advances, resembling AlphaFold, predict constructions from sequences however generate just a few conformations, lacking the dynamic nature of proteins. This limitation impacts docking accuracy, as AlphaFold-predicted constructions could not replicate the optimum configurations for ligand binding, resulting in inaccurate predictions.
Researchers from Galixir Applied sciences, Faculty of Pharmaceutical Science, Solar Yat-sen College, Heart for Theoretical Organic Physics and Division of Chemistry, Rice College, and World Institute of Future Expertise, Shanghai Jiao Tong College have developed DynamicBind, a deep studying technique that makes use of equivariant geometric diffusion networks to create a easy vitality panorama, enabling environment friendly transitions between totally different equilibrium states. DynamicBind precisely predicts ligand-specific conformations from unbound protein constructions with out holo-structures or intensive sampling. It excels in docking and digital screening benchmarks, accommodating giant protein conformational modifications, and figuring out hidden pockets in new protein targets. This technique reveals promise in accelerating the event of small molecules for beforehand undruggable targets, advancing computational drug discovery.
DynamicBind, a geometrical deep generative mannequin for dynamic docking, effectively adjusts protein conformations from preliminary AlphaFold predictions to holo-like states. It handles vital conformational modifications, just like the DFG-in to DFG-out transition in kinases, higher than conventional molecular dynamics simulations. DynamicBind achieves this by studying a funneled vitality panorama that minimizes frustration throughout transitions between biologically related states. Not like conventional Boltzmann mills, DynamicBind is generalizable to new proteins and ligands.
The DynamicBind mannequin is an E(3)-equivariant, diffusion-based graph neural community utilizing a coarse-grained illustration to foretell protein-ligand binding conformations. It effectively transforms enter constructions to account for 3D trans-rotational and parity modifications, outperforming conventional strategies with much less information. The mannequin employs a morph-like transformation for coaching, interpolating between crystal and AlphaFold constructions. Using a graph illustration, every protein residue and ligand atom is a node with numerous options. DynamicBind updates these nodes by tensor merchandise and diffusion processes to foretell side-chain dihedrals, torsion angles, translations, and rotations, enhancing binding affinity predictions.
DynamicBind is a flexible software for predicting protein-ligand advanced constructions, adept at accommodating vital protein conformational modifications. Throughout inference, it step by step adjusts ligand positions and inside angles over 20 iterations whereas additionally adapting protein conformations, notably side-chain angles, bettering upon AlphaFold-predicted constructions. Not like conventional fashions, it employs a morph-like transformation slightly than Gaussian noise perturbations, which boosts the mannequin’s potential to seize biologically related conformational modifications. DynamicBind excels in ligand pose prediction, decreasing clashes and revealing cryptic pockets, as demonstrated throughout numerous benchmarks and case research, showcasing its potential for drug discovery functions.
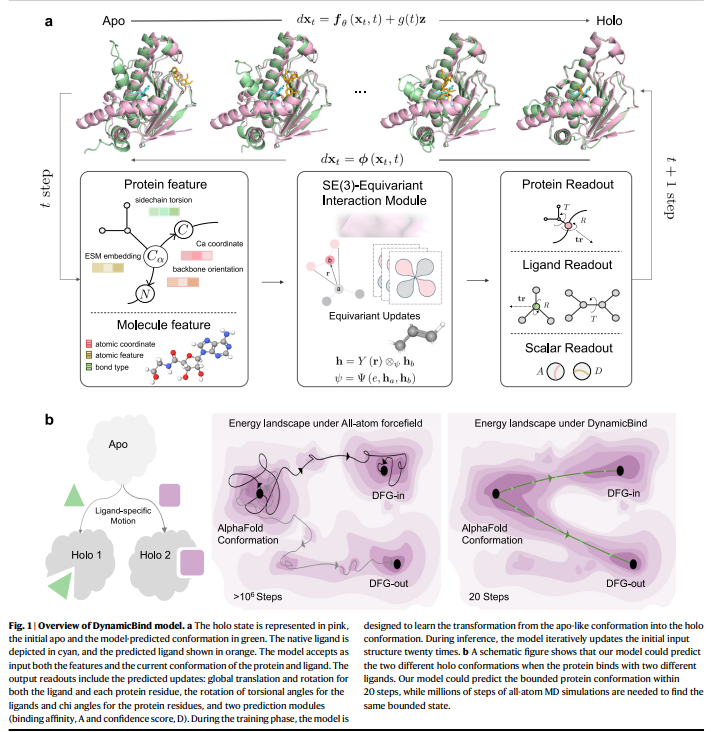
In conclusion, DynamicBind integrates protein conformation era and ligand pose prediction right into a single, end-to-end deep studying framework, considerably sooner than conventional MD simulations. Not like standard docking strategies that require predefined binding pockets, DynamicBind performs world docking, which is right for figuring out cryptic pockets. This reduces potential unintended effects by focusing on particular proteins and aids drug discovery by predicting unintended protein targets or figuring out targets in phenotype screening. Though it reveals glorious efficiency, enhancements are wanted for higher generalization to proteins with low sequence homology. Advances in Cryo-EM and incorporating binding affinity information can improve DynamicBind’s capabilities.
Take a look at the Paper and GitHub. All credit score for this analysis goes to the researchers of this undertaking. Additionally, don’t overlook to observe us on Twitter. Be part of our Telegram Channel, Discord Channel, and LinkedIn Group.
In case you like our work, you’ll love our publication..
Don’t Neglect to affix our 42k+ ML SubReddit
Sana Hassan, a consulting intern at Marktechpost and dual-degree scholar at IIT Madras, is keen about making use of expertise and AI to deal with real-world challenges. With a eager curiosity in fixing sensible issues, he brings a contemporary perspective to the intersection of AI and real-life options.
[ad_2]